

Les inspections menées par la FDA font partie intégrante du fonctionnement du marché américain. Leur déclenchement repose sur une combinaison de critères liés au produit, à l’organisation du fabricant et à l’environnement réglementaire dans lequel il évolue. Les entreprises qui commercialisent un dispositif médical aux États-Unis doivent disposer d’un système qualité en capacité d’expliquer, de documenter et de démontrer l’ensemble des décisions prises durant le cycle de vie du produit. Le niveau de rigueur attendu implique une organisation mature, des processus appliqués et une traçabilité maîtrisée. Le cas de DFI et de son dispositif médical Cybow 11 permet d’illustrer les écarts couramment observés lorsqu’un système qualité repose sur des principes formels sans application concrète. L’objectif de cet article est d’introduire les activités de la FDA en matière de surveillance.
La FDA applique un processus précis pour encadrer ses inspections de fabricants de dispositifs médicaux. Cette procédure s’appuie sur des critères prédéfinis pour organiser les visites, évaluer les systèmes qualité, et déterminer les suites à donner. La conformité est vérifiée au regard du 21 CFR Part 820, avec une attention particulière portée à la cohérence entre les procédures internes, les pratiques observées et les enregistrements disponibles.
La classe de risque du dispositif médical est le premier facteur de ciblage de la FDA.
Les dispositifs de classe III font l’objet de contrôles rapprochés. Une inspection peut avoir lieu avant l’approbation du PMA, puis à intervalles réguliers durant la phase de commercialisation. Les dispositifs de classe II sont souvent inspectés dans les deux premières années suivant leur mise sur le marché. Le cycle peut se poursuivre sur une fréquence de deux à trois ans, selon le profil de l’entreprise. Les produits de classe I sont inspectés à une fréquence moindre, sauf en cas de signalement.
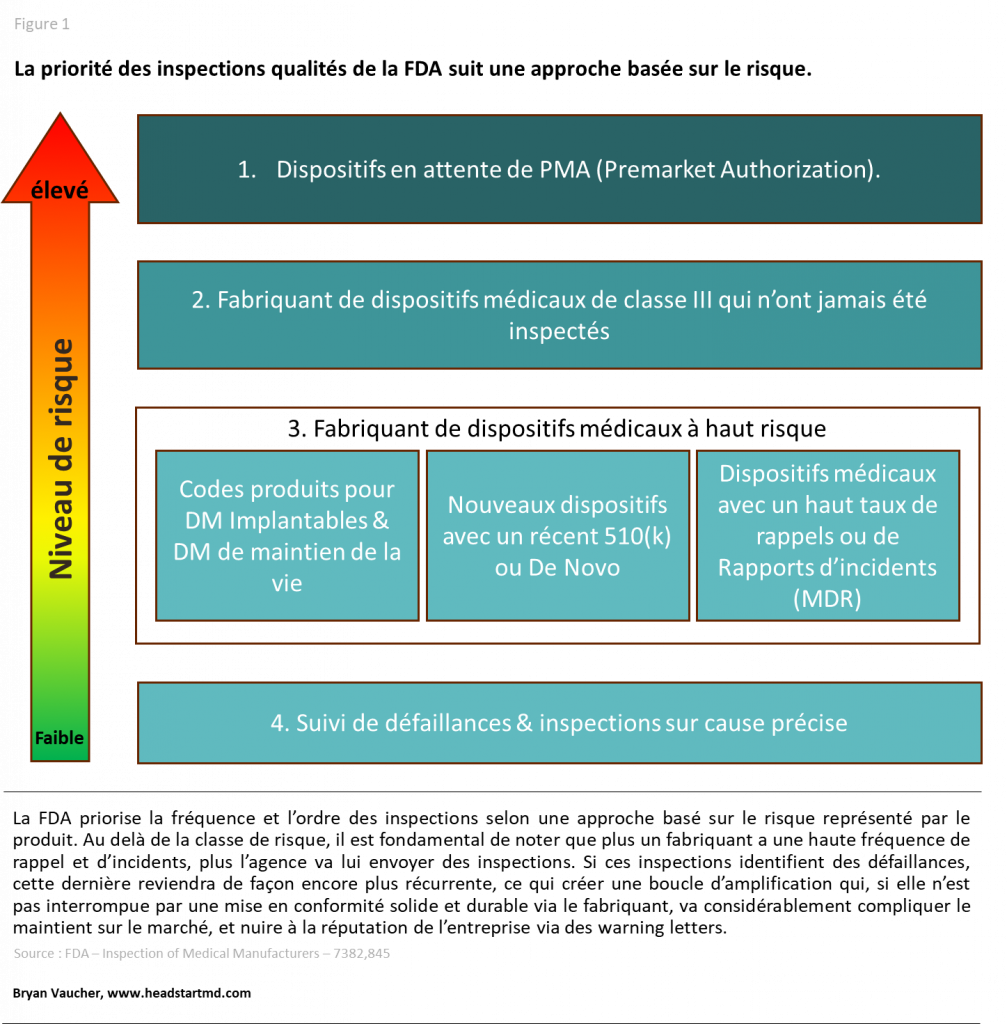
En addition au critère de sécurité, la FDA croise plusieurs types d’informations tels que des rappels récents, des réclamations enregistrées, des éléments de matériovigilance, mais aussi des critères relatifs aux spécificités du produit tels que le procédé stérilisation/reprocessing ou l’usage d’intelligence artificielle. L’inscription récente d’un établissement dans la base de données FDA, ou des incohérences dans les listings, constituent également des déclencheurs possibles.
Le délai de préavis peut varier. Certaines inspections sont annoncées cinq jours à l’avance, d’autres sont inopinées. Dans tous les cas, l’organisation doit être en toute circonstance en capacité de présenter un système opérationnel, accessible, cohérent et à jour.
L’objectif de l’inspecteur FDA est de vérifier que les exigences du 21 CFR Part 820 sont traduites dans les faits. Il examine si les processus sont documentés, compris, mis en œuvre de façon constante, et contrôlés. Il s’intéresse également à la manière dont les décisions sont prises, justifiées et tracées. L’analyse porte sur la robustesse du système qualité dans son ensemble. Chaque élément doit être démontrable à partir de preuves internes disponibles immédiatement.
Le cas de l’inspection de DFI pour Cybow 11
En janvier 2025, la FDA a mené une inspection dans l’établissement de DFI Co., fabricant du dispositif médical de diagnostic in vitro urinaire Cybow 11. La visite a conduit à l’émission d’une warning letter documentant une série d’écarts majeurs.
Certaines réclamations clients n’étaient pas enregistrées.
D’autres, bien qu’identifiées, étaient traitées de manière informelle, sans passage par le système qualité. Des tests contrôles qualités sur les réactifs étaient répétés suite à un échec sans justification structurée, sur la seule base de l’intuition d’un opérateur.
Les procédures de traitement des non-conformités n’étaient pas suivies. Le contrôle de l’étiquetage était incomplet.
Les DHR, censés tracer l’exécution du processus de fabrication du dispositif médical, ne contenaient pas les données techniques permettant de confirmer les étapes critiques. Enfin, la procédure de matériovigilance ne permettait pas d’identifier ni d’évaluer de manière fiable les événements potentiellement déclarables.
La FDA a également relevé que certaines mises à jour documentaires apportées en réponse à l’inspection ne modifiaient pas le comportement attendu. Les documents avaient été corrigés, mais leur application restait insuffisamment définie, ou sans mécanisme de vérification associé. La warning letter souligne l’absence de mesures correctives couvrant l’ensemble du portefeuille produit, alors que les constats dépassaient le seul périmètre du dispositif Cybow 11.
Ce cas montre que l’accumulation de procédures et de formulaires ne suffit pas. La FDA évalue la continuité du raisonnement qualité, la capacité à identifier, gérer, tracer et corriger les situations déviantes, sans dépendre d’initiatives individuelles non encadrées.
Une Warning Letter est une notification publique émise par la FDA lorsqu’elle identifie des manquements significatifs au cadre réglementaire lors d’une inspection et que la réponse écrite du fabriquant au rapport d’inspection est insuffisante.
Ce courrier vise à formaliser les écarts constatés, en exigeant une réponse documentée dans un délai précis. Il est publié en ligne par l’agence, accessible à tous les acteurs du secteur, y compris les investisseurs, partenaires industriels, autorités de santé étrangères, clients ou distributeurs.
Pour le fabriquant du dispositif médical concerné, ce type de publication génère un préjudice d’image immédiat. Il affecte la crédibilité de l’organisation, fragilise les négociations commerciales en cours, et peut déclencher un effet de propagation réglementaire, notamment dans les pays ayant signé des accords de reconnaissance mutuelle.
Dans certains cas, la réception d’une warning letter entraîne également la suspension des dépôts réglementaires en cours ou une restriction temporaire de commercialisation.
Au-delà de l’impact opérationnel, une warning letter est souvent perçue comme un signal fort de gouvernance insuffisante.
Se préparer à une inspection FDA ne consiste pas à mobiliser des ressources à la veille d’une visite. Le système qualité doit être conçu pour fonctionner en régime continu. L’enjeu n’est pas de répondre à des questions, mais d’opérer selon une logique de justification structurée à chaque étape du développement, de la fabrication et de la distribution de votre dispositif médical.
Cette exigence transforme la manière dont les dirigeants doivent aborder la gouvernance des opérations. Les procédures ne sont pas rédigées pour répondre à un besoin externe. Elles permettent de guider, de tracer, et de maintenir la cohérence de l’ensemble du système et donc de a la qualité du produit fini. Leur présence n’a de valeur que si elle est associée à des mécanismes de preuve, à des arbitrages formalisés, et à des décisions alignées avec les exigences réglementaires.
La capacité à maintenir un niveau de maîtrise stable est aujourd’hui considérée par la FDA comme un facteur de légitimité dans la durée. Cette stabilité constitue également un levier de confiance pour les financeurs, les distributeurs et les partenaires industriels et hospitaliers. Elle influence la perception du projet, sa valorisation, et son potentiel de développement.